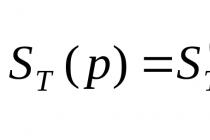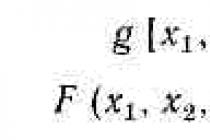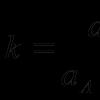^ Maticová metoda
Kromě metody orientovaného grafu existují další metody řešení stechiometrických úloh pro složité systémy chemických reakcí. Maticová metoda umožňuje redukovat problém do podoby, která je nejvhodnější pro jeho další řešení pomocí výpočetní techniky.
Uvažujme řešení předchozího problému pomocí maticové metody. 7 látek se účastní systému 4 chemických reakcí. Rovnice chemických reakcí zahrnujících tyto látky lze zapsat tak, jako by se jich všechny látky účastnily současně. Pokud se látka neúčastní nějaké chemické reakce, formálně to znamená, že stechiometrický koeficient této látky je nulový. Souhlasíme také s tím, že stechiometrické koeficienty pro výchozí látky budou považovány za kladné a pro produkty - záporné. Pak lze první z chemických rovnic systému chemických reakcí uvažovaných v předchozím příkladu zapsat takto:
A + 2B - 2C + 0D + 0E + 0F + 0H = 0.
Pokud budeme argumentovat podobně pro všechny látky a všechny reakce, sestavíme soustavu lineárních rovnic popisujících poměr hmotností všech látek účastnících se reakcí. Rozměr soustavy je 4x7, kde 4 je počet rovnic, 7 je počet látek účastnících se chemických reakcí. Matice koeficientů těchto rovnic je uvedena níže a sloupcový vektor je nulový.
K výsledné soustavě rovnic je potřeba přidat ještě několik rovnic, které mají nenulovou pravou stranu. Tyto rovnice jsou napsány na základě počátečních podmínek problému.
A B C D E F H
1 2 -2 0 0 0 0 0
1 0 0 -2 0 0 0 0
0 0 1 -1 0 -1 0 0
0 0 0 1 0 -2 -1 0
Za určitých podmínek, kdy jsou známy hodnoty počáteční a aktuální hmotnosti některých složek systému, je možné získat jediné řešení metodami lineární algebry.
Popis soustav výpočtem stechiometrie chemických reakcí z praktického hlediska umožňuje vypočítat hmotnosti všech zúčastněných látek. Lze tak předvídat chování systému, složení produktů, množství spotřebovaných látek.
Stechiometrické výpočty předpokládají, že všechny chemické reakce v daném technologickém procesu probíhají zcela doprava.
^ Modelování rovnováhy v systémech chemických reakcí
Významná část chemických reakcí, které tvoří hlavní náplň technologických procesů v metalurgii neželezných kovů, je vratná. Zvažte příklad reverzibilní chemické reakce:
Rovnováhy v takové chemické reakci je dosaženo při určitých hodnotách aktivit zúčastněných látek. Pokud jsou tyto látky v roztoku a jejich koncentrace jsou nízké (zředěné roztoky), lze s určitou aproximací místo hodnot aktivity použít hodnoty koncentrace. Rovnováhu v chemické reakci charakterizuje hodnota rovnovážné konstanty:
 .
.
Hodnota rovnovážné konstanty je spojena se změnou Gibbsovy energie a lze ji vypočítat z termodynamických dat zúčastněných látek:

kde Δ G T- změna Gibbsovy energie pro danou chemickou reakci, T- teplota, R je univerzální plynová konstanta.
Po výpočtu hodnoty rovnovážné konstanty pro chemickou reakci probíhající při dané teplotě je možné určit poměr koncentrací výchozích látek a produktů, které nastanou při dosažení rovnováhy.
Poněkud obtížnější je určit rovnovážné složení systému, ve kterém probíhá několik vratných chemických reakcí současně. Zvažte následující příklad. Nechť existuje systém vratných chemických reakcí zahrnujících látky A, B, C a D. V tomto systému se látka A postupně a reverzibilně přeměňuje na látku C, předběžně tvoří B. Je možná i paralelní cesta: látka A, v souběžně se vznikem B se rozkládá na formu D. Za daných podmínek (teplota, tlak) bude v systému nastolena rovnováha a rovnovážné koncentrace látek.
Pro výpočet rovnovážných koncentrací zapíšeme výrazy pro rovnovážné konstanty všech reakcí z hlediska rovnovážných koncentrací:
A B  ;
; 
PŘED NAŠÍM LETOPOČTEM  ;
;
A D  ;
;  .
.
Nechť v počátečním okamžiku neexistují žádné meziprodukty B a C, stejně jako konečný produkt D:
 ; CBO = 0; С С0 = 0; CDo = 0.
; CBO = 0; С С0 = 0; CDo = 0.
Hodnoty rovnovážných konstant se vypočítají pro každou z reakcí pomocí termodynamických dat:  ... Hodnoty rovnovážných konstant tedy budou považovány za známé hodnoty.
... Hodnoty rovnovážných konstant tedy budou považovány za známé hodnoty.
Na jednotku objemu tohoto systému je C A0 - CA počet spotřebovaných molů složky A. V souladu se stechiometrií chemických reakcí a zákonem zachování hmoty je ztráta hmotnosti A rovna součtu hmotností. výsledných látek B, C a D, které lze vyjádřit rovnicí:
CA0 - CA = C B + C C + C D.
Převedeme rovnici do následujícího tvaru:
C A0 = C A + C B + C C + C D,
A dosadíme na pravou stranu výrazu odpovídající koncentrace látek:
C A0 = C A + k 1 C A + k 1 k 2 C A + k 3 C A.
Seskupujeme homogenní členy rovnice
CA 0 = CA (1 + k 1 + k 1 k 2 + k 3)
a získáme výraz pro rovnovážnou koncentraci C A
 .
.
Rovnovážné koncentrace ostatních látek lze snadno určit, protože hodnoty všech rovnovážných konstant jsou nám známy z předchozího výpočtu a výrazy obsahují C A.
Při výpočtu rovnováh v systémech chemických reakcí je nutné znát k p každé reakce, počáteční složení systému - to umožňuje vypočítat rovnovážné složení systému.
Skutečné problémy výpočtu rovnovážného složení systémů jsou mnohem složitější: rovnice v těchto úlohách jsou nelineární; je třeba vzít v úvahu, že složky vstupující do reakce jsou v různých fázích; místo koncentrací je správné použít hodnoty aktivit složek. Praktický význam výpočtu rovnováh v takto složitých systémech se scvrkává na skutečnost, že vypočítané rovnovážné složení systému je fyzikálně-chemická mez, které může skutečný proces dosáhnout, pokud je pro jeho realizaci vyhrazena neomezená doba.
^ Modelování kinetiky chemických reakcí
Ve fyzikální chemii se rychlost chemické reakce určuje podle rovnice:
 ,
,
kde dq- změna hmotnosti reaktantu, mol.
dt- přírůstek času, s.
PROTI Je mírou reakčního prostoru.
Rozlišujte mezi homogenními chemickými reakcemi, ve kterých jsou všechny zúčastněné látky ve stejné fázi (plyn nebo kapalina). Pro takové reakce je mírou reakčního prostoru objem a rozměr rychlosti bude:  .
.
Mezi látkami v různých fázích (plyn-pevná látka, plyn-kapalina, kapalina-kapalina, pevná látka-kapalina) probíhají heterogenní chemické reakce. V tomto případě je skutečná chemická reakce realizována na rozhraní, které je mírou reakčního prostoru.
U heterogenních reakcí je rozměr rychlosti jiný:  .
.
Změna hmotnosti reagujících látek má své vlastní znamení. U výchozích látek hmotnost v průběhu reakce klesá, změna hmotnosti má záporné znaménko a velikost rychlosti nabývá záporné hodnoty. U produktů chemické reakce se hmotnost zvětšuje, změna hmotnosti je kladná a kladné se předpokládá i znaménko rychlosti.
Zvažte jednoduchou chemickou reakci
A + 2B = 2C.
Mezi jednoduché reakce patří ty, které se provádějí v jedné fázi a jdou do konce, tzn. jsou nevratné.
Pojďme určit rychlost takové chemické reakce. K tomu je nejprve nutné rozhodnout, pro kterou z látek bude rychlost reakce stanovena: vždyť A a B jsou výchozí látky a změna jejich hmotnosti je záporná a C je konečný produkt, a jeho hmotnost s časem roste. Kromě toho, ne všechny stechiometrické koeficienty v reakci jsou rovny jednotce, což znamená, že pokud je průtok A po určitou dobu roven 1 molu, průtok B za stejnou dobu bude 2 moly, a proto hodnoty rychlosti vypočítané ze změny hmotností A a B se budou lišit o polovinu.
Pro jednoduchou chemickou reakci lze navrhnout jedinou míru rychlosti, která se určí takto:
 ,
,
kde r i- rychlost i-tého účastníka reakce
S i Je stechiometrický koeficient i-tého účastníka reakce.
Stechiometrické koeficienty pro výchozí látky jsou považovány za kladné, pro reakční produkty jsou záporné.
Pokud reakce probíhají v izolované soustavě, která nevyměňuje hmotu s vnějším prostředím, pak pouze chemická reakce vede ke změně hmotností hmoty v soustavě a následně i jejich koncentrací. V takovém systému je jediným důvodem změny koncentrace S je chemická reakce. Pro tento konkrétní případ
 ,
,
Rychlost chemické reakce závisí na koncentracích obsažených látek a na teplotě.

kde k - rychlostní konstanta chemické reakce, S A ,S PROTI- koncentrace látek, n 1 , n 2 - objednávky příslušných látek. Tento výraz je ve fyzikální chemii známý jako zákon hromadného působení.
Čím vyšší jsou hodnoty koncentrace, tím vyšší je rychlost chemické reakce.
Objednat ( n) je stanoven experimentálně a je spojen s mechanismem chemické reakce. Pořadí může být celé číslo nebo zlomkové číslo, u některých látek existují i reakce nultého řádu. Pokud je objednávka i-tá látka je rovna nule, pak rychlost chemické reakce nezávisí na koncentraci této látky.
Rychlost chemické reakce závisí na teplotě. V souladu s Arrheniovým zákonem se rychlostní konstanta mění se změnou teploty:

kde ^ A- preexponenciální faktor;
E- aktivační energie;
R- univerzální plynová konstanta, konstantní;
T- teplota.
Stejně jako velikost řádu reakce jsou pro konkrétní reakci experimentálně určeny hodnoty aktivační energie a preexponenciálního faktoru.
Pokud chemická reakce probíhá v heterogenním procesu, pak je její rychlost ovlivněna i procesem dodávání výchozích látek a odstraňování produktů ze zóny chemické reakce. Probíhá tedy složitý proces, ve kterém dochází k difúzním fázím (přísun, odběr) a kinetické fázi – vlastní chemické reakci. Rychlost celého procesu jako celku, pozorovaného v experimentu, je určena rychlostí nejpomalejšího stupně.
Ovlivněním rychlosti difúzního stupně procesu (míchání) tedy ovlivňujeme rychlost celého procesu jako celku. Tento vliv ovlivňuje hodnotu preexponenciálního faktoru A.
Většina chemických reakcí není jednoduchá (to znamená, že nejdou v jedné fázi a ne do konce) - složité chemické reakce:
A) AB - reverzibilní;
B) A -> B; В → С - po sobě jdoucí;
B) A -> B; A → C - paralelní.
Pro složitou chemickou reakci neexistuje jednotná míra rychlosti... Na rozdíl od jednoduchého zde můžeme mluvit o rychlosti tvorby a ničení každé chemikálie. Pokud tedy v systému probíhají chemické reakce a n látky pro každou z n látky mají svou vlastní hodnotu rychlosti.
Pro kteroukoli z látek je rychlost tvorby a zániku algebraickým součtem rychlostí všech stupňů za účasti této látky.
Rychlost komplexní chemické reakce
Uvažujme modelování kinetiky systému složitých chemických reakcí pomocí následujícího příkladu. Nechť existuje technologický proces, jehož podstata se projevuje následujícími chemickými reakcemi:
K1; 1 až B
K2; 0,7 až C
K 3; 1 až A; 0,35 v N
K 4; 1 až C; 1 až D
K 5; 2 až E;
RA = –k 1 C B + k 2 C C 0,7 - k 3 C A CH 0,35
R B= –2k 1 C B + 2k 2 C C 0,7
R C = k 1 C B - k 2 C C 0,7 - k 4 C C C D + k 5 C E 2
R D = k 3 C A CH 0,35 - k 4 C C C D + k 5 C E 2
RE = k 4 C C C D - 3k 5 C E 2
Kinetické konstanty (řády látek a hodnoty rychlostních konstant pro stadia) byly stanoveny experimentálně. V procesním diagramu nad šipkami odpovídajícími stupňům jsou znázorněny velikosti řádů látek. Nespecifikované objednávky jsou nulové.
Procesu se účastní 6 látek: A a B jsou počáteční, C a D jsou meziprodukty, E je konečný produkt, H je katalyzátor jednoho ze stupňů. Tři chemické reakce mají pět stupňů, z nichž tři jsou přímé a dvě jsou reverzní.
Všechny reakce probíhají homogenně a probíhají v hmotně uzavřeném systému, což dává důvod použít k charakterizaci rychlosti následující výrazy:
 .
.
Na základě výše uvedeného si zapíšeme výrazy pro rychlosti pro každou látku-účastníka. Celkem dostaneme 6 výrazů pro počet látek. Pro každou z látek je rychlost spotřeby nebo tvorby algebraickým součtem rychlostí všech stupňů za účasti dané látky. Látka A se tedy účastní tří stupňů, v prvním jako výchozí látka, ve druhém jako produkt, ve třetím opět jako výchozí látka. Rychlostní členy pro první a třetí stupeň budou záporné, pro druhý stupeň má rychlost kladné znaménko. Hodnoty rychlosti pro každý stupeň jsou podle zákona efektivních hmotností součinem rychlostní konstanty příslušného stupně a koncentrací látek v mocninách rovných řádům látek. S ohledem na to budou výrazy pro rychlosti látek následující:
 = –K 1 C B + k 2 C C 0,7 - k 3 C A CH 0,35
= –K 1 C B + k 2 C C 0,7 - k 3 C A CH 0,35
 = –2k 1 C B + 2k 2 C C 0,7
= –2k 1 C B + 2k 2 C C 0,7
 = k 1 C B - k 2 C C 0,7 - k 4 C C C D + k 5 C E 2
= k 1 C B - k 2 C C 0,7 - k 4 C C C D + k 5 C E 2
 = k 3 C A CH 0,35 - k 4 C C C D + k 5 C E 2
= k 3 C A CH 0,35 - k 4 C C C D + k 5 C E 2
 = 3k 4 C C C D - 3k 5 C E 2
= 3k 4 C C C D - 3k 5 C E 2
 = 0.
= 0.
Poslední rychlost pro látku H, katalyzátor třetího stupně, je nula. Hmotnost katalyzátoru se v průběhu reakce nemění.
Na levé straně všech rovnic je derivace koncentrace hmoty v závislosti na čase, proto jsou rovnice kinetiky diferenciální. Koncentrace na pravé straně rovnic v libovolném časovém okamžiku musí současně splňovat všechny rovnice, což znamená, že soubor kinetických rovnic v matematickém smyslu je soustavou rovnic.
Chemický kinetický model je soustava diferenciálních rovnic, jejichž řešením je množina funkcí C i = F i (t) :
CA = f 1 (t)
Pro stanovení konkrétního tvaru funkcí je nutné řešit soustavu diferenciálních rovnic, tzn. integrovat systém kinetických rovnic. Níže zvážíme integraci rovnic kinetiky pomocí jednoduššího příkladu a poté se vrátíme k výše uvedenému problému.
^ Integrace kinetických rovnic
Nechť dojde k chemické reakci rozkladu látky A, v jejímž důsledku vzniká látka B. Experimentálně bylo zjištěno, že má první řád v koncentraci A a hodnota rychlostní konstanty pro podmínky její realizace se rovná k. To je znázorněno v reakčním diagramu níže.
k; 1 až A
Reakční rychlost je r a = –kC A, nebo
 .
.
Definujme počáteční podmínky pro řešení diferenciální rovnice kinetiky. Budeme předpokládat, že v počátečním okamžiku reakce známe koncentraci látky A, označíme ji jako C Ao. Počáteční podmínky zapíšeme do formuláře  ... Výslednou rovnici budeme integrovat pomocí integrálů s dosazením limit. Meze integrace jsou určeny z počátečních podmínek: když je čas nula, koncentrace A je rovna počáteční, v libovolném okamžiku t koncentrace je S A :
... Výslednou rovnici budeme integrovat pomocí integrálů s dosazením limit. Meze integrace jsou určeny z počátečních podmínek: když je čas nula, koncentrace A je rovna počáteční, v libovolném okamžiku t koncentrace je S A :
 .
.
V důsledku integrace máme:
 ,
,
Nahrazením rozdílu logaritmů logaritmem kvocientu máme další:
 ,
,
Provedením potenciace získáme:
 .
.
Po všech transformacích je řešením diferenciální rovnice exponenciální klesající funkce:
 .
.
Zkontrolujme, zda získané řešení odporuje podmínkám našeho problému. Na t= 0, tj. na začátku chemické reakce S A = C A 0 protože exponent se stane jedničkou. Ve skutečnosti je v počátečním okamžiku koncentrace látky A rovna počáteční. Na t→ ∞ exponent se záporným exponentem má tendenci k nule. Během nekonečně dlouhého času se veškerá hmota v důsledku chemické reakce rozloží a vytvoří V.
^ Metody numerické integrace
Vraťme se nyní k předchozímu problému. Je zřejmé, že integrace systému diferenciálních rovnic je ve srovnání s dříve uvažovaným úkolem obtížnější. Použití analytických metod integrace je stěží možné, protože pravé strany diferenciálních rovnic obsahují koncentrace několika látek najednou a nebude možné proměnné oddělit.
Použijme metodu numerické integrace. K tomu rozdělíme časovou osu na malé segmenty (kroky). Za předpokladu, že časové derivace koncentrací látek jsou matematickým limitem poměru přírůstků koncentrace k přírůstku času, při Δ t , sklon k nule:
 ,
,
transformujeme soustavu diferenciálních rovnic na soustavu algebraických. Na levé straně známe časový přírůstek, protože časový krok volíme sami. Důležité je pouze to, aby tento krok byl malý.
Na pravé straně také známe hodnoty všech rychlostních konstant z experimentu, totéž je třeba říci o velikostech řádů. Dosadíme také hodnoty koncentrací všech látek na pravé straně pomocí počátečních podmínek. Každá z rovnic soustavy obsahuje v tomto případě pouze jednu neznámou veličinu - změnu koncentrace Δ C i... Ve skutečnosti se jedná o změnu koncentrace pro první krok roztoku, kdy se čas změní z nuly (začátek chemické reakce) do Δ t. Změna koncentrace s vlastním znaménkem se sečte s počáteční koncentrací a stanoví se koncentrace každé z látek na konci prvního kroku roztoku.
V dalším kroku řešení dosadíme hodnoty koncentrace z předchozího kroku řešení na pravou stranu a opět získáme Δ C i ale nyní k dalšímu kroku řešení, jak je znázorněno na obrázku.
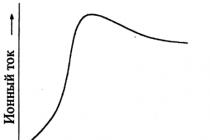
V každém kroku řešení získáme pořadnice odpovídající změně koncentrace všech látek účastnících se reakcí. Umístění bodů, které jsou pořadnicemi, poskytne pro každou látku graf funkce změny koncentrace v čase. Všimněte si, že v důsledku numerické integrace nezískáme analytický výraz, který specifikuje změnu koncentrace v čase, souřadnice na grafu jsou získány výpočtem. Konstrukce grafů funkcí změn koncentrace v čase je však možná a tvar křivek nám umožňuje vyvodit řadu závěrů, které mají praktický význam.
Je zřejmé, že koncentrace výchozích látek s časem klesají, protože se při reakci spotřebovávají. Je neméně zřejmé, že koncentrace konečných produktů se zvyšují.
Samostatnou úvahu si zaslouží chování meziproduktů. Grafy koncentrací meziproduktů mají maxima odpovídající určité reakční době. Pokud je meziprodukt cílovým produktem chemických reakcí, pak maximální koncentrace odpovídá optimální době trvání pro získání této cílové látky.
V počátečním okamžiku chemické reakce jsou totiž koncentrace výchozích látek vysoké a rychlost chemické reakce za účasti výchozích látek je úměrná jejich koncentracím. Reakce zahrnující výchozí materiály zpočátku probíhají vysokou rychlostí. To znamená, že meziprodukty vznikají také vysokou rychlostí.
Na druhou stranu rychlost rozkladu meziproduktů je také úměrná jejich koncentraci a je zpočátku malá. Rychlost tvorby meziproduktů je větší než rychlost jejich rozkladu, což přispívá k akumulaci meziproduktů, zvyšuje se jejich koncentrace.
S rozvojem chemické reakce klesá rychlost tvorby meziproduktů a zvyšuje se rychlost jejich destrukce. Když se hodnoty rychlostí stanou stejnými, nárůst koncentrace se zastaví a v systému je pozorována maximální koncentrace meziproduktu.
Dále se rychlost tvorby meziproduktu snižuje s tím, jak se koncentrace výchozích materiálů dále snižuje. Snižuje se také rychlost destrukce mezilátky, která zůstává větší než rychlost tvorby, což vede ke spotřebě mezilátky v systému a k poklesu její koncentrace.

 ;
;
T C je optimální doba pro získání látky C.
Uvažujme chování látky C: v počátečním okamžiku C C = 0.
K 1 C B > k 2 C C 0,7
Modelování kinetiky tak umožňuje určit vznik a spotřebu všech látek v systémech chemických reakcí, stanovit formu koncentrační funkce jako funkci času a v některých případech určit optimální podmínky pro vedení chemické látky. reakce.
^ Chemické reakce v toku látek
Mnoho technologických zařízení pracuje v nepřetržitém režimu. Uvažujme jako příklad tavicí pec pro zpracování vsázky z měděných koncentrátů a tavidel. Schéma takového zařízení je znázorněno níže na obrázku.
H  Kontinuální průtokové zařízení je průtokový reaktor, ve kterém se provádí určitá sada chemických reakcí.
Kontinuální průtokové zařízení je průtokový reaktor, ve kterém se provádí určitá sada chemických reakcí.
Přítomnost toků látek ovlivňuje podmínky pro provádění chemických reakcí.
Skutečné toky hmoty mají poměrně složité vlastnosti:
hydrodynamický režim - laminární, turbulentní, přechodný;
počet fází - více- a jednofázové.
Příkladem je proud pohybující se potrubím. Rychlost proudění v rámci jednoho úseku není stejná: nejvyšší hodnota rychlosti je na ose proudění a u stěn se vlivem zpomalování proudění silami viskozity tato rychlost jen málo liší od nuly. . Pokud je však objemový průtok průtokového média Q a plocha průřezu F, je snadné určit průměrný průtok průtoku rovný Q / F.
 Q m3/s
Q m3/s

Ještě větší potíže vyvstávají při popisu vícefázového proudění, přičemž skutečnými proudy jsou nejčastěji.
V tomto ohledu je poměrně obtížné při vytváření matematického modelu zohledňovat vlastnosti skutečných toků. Proto pro vytvoření modelu průtokového aparátu existuje několik idealizovaných modelů toku toků.
^ 1. Model plug-flow - takový idealizovaný model toku je založen na následujících předpokladech (tímto typem zařízení by mohla být trubková pec):
průtok je stacionární, objemový průtok média se v čase nemění;

v takovém proudu jsou rychlosti ve všech bodech proudu stejné;
prvek objemu dV v takovém toku je hmotně uzavřený systém (nevyměňuje se se sousedními prvky);
v pístovém toku nedochází k podélnému míchání;
nedochází také k žádnému křížovému míšení v proudu.
Pro modelování kinetiky v případě pístového toku je docela vhodný přístup aplikovatelný na systémy izolované v hmotě.
Uvažujme reakci prvního řádu, která probíhá v posuvném aparátu.
K1; 1 až A
S  Vytvořme model, který nám umožní vypočítat výstupní koncentraci A. Konstanta je známá, prvního řádu.
Vytvořme model, který nám umožní vypočítat výstupní koncentraci A. Konstanta je známá, prvního řádu.



 - doba setrvání látky v přístroji
- doba setrvání látky v přístroji


Čím větší je rychlostní konstanta k tím rychleji má koncentrace tendenci ke koncentraci na výstupním bodě.
Koncentrace látky nezůstává ve vytěsňovacím aparátu konstantní - klesá z koncentrace na vstupním bodě na koncentraci na výstupním bodě.
^ 2. Model dokonalého promíchání (zařízením tohoto typu je např. KS pec, hydrometalurgický louhovací reaktor aj.).
Předpoklady:
průtok je stacionární, objemový průtok látky (Q) zařízením musí být konstantní;
koncentrace ve všech bodech ideálního míchacího zařízení je stejná.
 Důsledkem druhého předpokladu je, že koncentrace látky na výstupu je rovna koncentraci uvnitř přístroje.
Důsledkem druhého předpokladu je, že koncentrace látky na výstupu je rovna koncentraci uvnitř přístroje. 
Průměrná doba setrvání látky v přístroji -  .
.
Doba zdržení různých částí proudu v ideálním míchacím zařízení není stejná.
Objemovým prvkem v takovém zařízení je otevřený systém, pro takové zařízení není přístup pro uzavřený systém vhodný. K popisu kinetiky v tomto případě použijeme zákon hmoty a uvažujeme aparaturu jako celek, koncentrace ve všech bodech je stejná. Na základě zákona zachování hmoty zapíšeme rovnici materiálové bilance pro celý aparát jako celek (za jednotku času):
Příjem – spotřeba = 0
Nechť proběhne rozkladná reakce prvního řádu za podmínek ideálního míchacího zařízení:
K1; 1 až A
Materiálová bilance pro látku A bude součtem složek:
1 člen - počet molů látky A vnesených tokem za jednotku času;
2 termín - strhávání hmoty z přístroje za jednotku času;
3 člen - hmotnost látky spotřebované při chemické reakci. Obě strany rovnice vydělíme objemovým průtokem Q ≠ 0:

 .
.
Vytvořme stejné podmínky pro chemické reakce v obou aparátech (stejná teplota,  k 1 = k 2). Předpokládejme, že při určité teplotě k 1 = k 2 = 1. sada CA0 = 1 mol/m3. Vа = 1 m 3, Q 1 = Q 2 = 1 m 3 / s. Pak:
k 1 = k 2). Předpokládejme, že při určité teplotě k 1 = k 2 = 1. sada CA0 = 1 mol/m3. Vа = 1 m 3, Q 1 = Q 2 = 1 m 3 / s. Pak:

 .
.
Výsledek stejné chemické reakce se překvapivě v různých zařízeních liší. Účinnější je zařízení s pístovým tokem, ve kterém je výstupní koncentrace nižší.
Důvodem není rychlost chemické reakce (ta je u obou zařízení stejná), ale přítomnost nebo nepřítomnost míšení prvků proudění. V zařízení ideálního míchání se koncentrace ustaví na výstupu, což je výsledek míchání částí látky, které byly uvnitř zařízení po různou dobu. Některé části látky rychle přeskakují aparaturu a reakční doba v takových částech je krátká, zatímco koncentrace látky A je naopak vysoká. Ostatní části látky zůstávají uvnitř zařízení po dlouhou dobu, doba trvání chemické reakce je dlouhá a zbytková koncentrace A je malá.
^ Model buněčného proudění ... Podle tohoto modelu je skutečný technologický aparát nahrazen idealizovaným schématem - sekvencí buněk ideálního míchání.
 1; 1 až A
1; 1 až A NS  ústa n = 2, pak na výstupu 1. buňky:
ústa n = 2, pak na výstupu 1. buňky: 

Pokud n buněk, pak 
Vezmeme-li v úvahu, že  - přejdeme k řešení ideálního posuvného aparátu. Pro n = 1 máme zřejmé řešení ideální míchací aparatury.
- přejdeme k řešení ideálního posuvného aparátu. Pro n = 1 máme zřejmé řešení ideální míchací aparatury.
Ukažme si na grafech, jak nám zvýšení počtu článků může umožnit pomocí buněčného modelu přejít od ideálního míchacího aparátu k dokonalému vytěsňovacímu aparátu.

H  Aby se vyloučilo podélné míšení v proudu, je pracovní objem zařízení rozdělen na části.
Aby se vyloučilo podélné míšení v proudu, je pracovní objem zařízení rozdělen na části.
Využívá se také kaskádování aparátů - sériové zapojení technologických aparátů pro vyrovnání výsledků chemických reakcí.
Modelování kinetiky v tocích chemických reakcí umožňuje s přihlédnutím k charakteristikám toku vypočítat charakteristiky zařízení (výstupní složení).
Katedra fyzikální chemie
Abramenkov A.V.
KINET
Program pro numerickou simulaci kinetiky
složité chemické reakce
Program KINET je určen k řešení přímých a inverzních kinetických problémů. Jako výchozí údaj je nastaveno kinetické schéma (mechanismus) procesu ve formě sady jednoduchých reakcí s uvedením rychlostních konstant a reakční rovnice lze zapsat ve formě blízké obvyklému chemickému zápisu. Kromě toho jsou uvedeny podmínky procesu - počáteční koncentrace činidel a teplota, stejně jako časový interval, během kterého má být roztok získán. V případě inverzního kinetického problému je také nutné specifikovat experimentální kinetické křivky.
Program samostatně sestaví soustavu diferenciálních rovnic a integruje ji. Výsledky jsou prezentovány numericky a graficky a lze je exportovat pro použití v jiných programech.
Některé příklady aplikace programu KINET jsou uvedeny v knize: „Praktická práce z fyzikální chemie: Kinetika a katalýza. Elektrochemie "(Abramenkov A.V., Ageev E.P., Atyaksheva L.F. et al. Editoval V.V. Lunin a E.P. Ageev). Moskva: Ed. centrum "Akademie", 2012. Sekce I.8, "Matematické modelování kinetiky komplexních reakcí", s. 70-102. Podrobný návod k používání programu naleznete v souboru UserGuide.pdf.
Požadavky na systém:
- Windows XP / Vista / 7 (32bitový nebo 64bitový),
- 3,7 MB na disku nebo flash disku pro umístění souborů programu,
- rozlišení obrazovky alespoň 1024 x 768 (raději vyšší).
Pokyny pro instalaci
Chcete-li nainstalovat program KINET, stačí rozbalit stažený archiv (zachování vnitřní struktury podsložek) do složky "Program Files" nebo jakékoli jiné složky a vytvořit na ploše zástupce pro spuštění spustitelného souboru wkinet.exe.
Uvnitř složky Kinet obsahuje podsložka locale překlad rozhraní programu do ruštiny. Bez této podsložky zůstane program funkční, ale bude mít anglické rozhraní.
Jednotlivá nastavení se ukládají do souboru kinet.ini ve standardní složce pro ukládání dat v uživatelském profilu. V moderních verzích Windows je to obvykle složka C: \ Users \<Имя пользователя>\ Aplikační data \ Kinet \
Program nic nezapisuje do registru Windows, takže pro jeho úplné odstranění z počítače stačí smazat hlavní složku Kinet s programovými soubory a složku Kinet v uživatelském profilu (viz výše).
Program KINET je šířen volně (viz licenční ujednání v souboru Kinet \ doc \ license_ru.txt).
Kroky simulace
Proces teoretického i experimentálního modelování se skládá z následujících kroků:
1. Stavba modelu.
2. Prostudujte si model.
3. Extrapolace - přenos získaných dat do oblasti znalostí o původním objektu.
V první fázi, po uvědomění si nemožnosti či nevhodnosti přímého studia objektu, je vytvořen jeho model. Účelem této etapy je vytvořit podmínky pro úplné nahrazení originálu zprostředkujícím objektem, který reprodukuje jeho potřebné parametry.
Ve druhé fázi se provádí studium samotného modelu - tak podrobné, jak je vyžadováno pro řešení konkrétního kognitivního úkolu. Výzkumník zde může pozorovat chování modelu, provádět na něm experimenty, měřit nebo popisovat jeho charakteristiky – v závislosti na specifikách samotného modelu a na počátečním kognitivním úkolu. Účelem druhé fáze je získat požadované informace o modelu.
Třetí etapa (extrapolace) je „návrat“ k původnímu objektu, tzn. interpretace získaných poznatků o modelu, posouzení jejich přijatelnosti a podle toho jejich aplikace na originál, umožňující v případě úspěchu vyřešit původní kognitivní problém.
Tyto kroky implementují jakýsi simulační cyklus, během kterého spolu model a originál spolu souvisí. (Obr. 1).
Rýže. 1. Kroky simulace
Simulace v chemii
Modelování molekul, chemických procesů a reakcí
Materiálové (experimentální) modelování je v chemii široce využíváno k pochopení a studiu struktury látek a charakteristik průběhu chemických reakcí, k identifikaci optimálních podmínek pro chemicko technologické procesy atd.
V biochemii a farmakologii modelování hraje velmi důležitou roli. Pokrok farmakologie je charakterizován neustálým hledáním a tvorbou nových, pokročilejších léků. V posledních letech se při vytváření nových léků nebere jako základ biologicky aktivní látka, jak tomu bylo dříve, ale substrát, se kterým interaguje (receptor, enzym atd.). Takové studie vyžadují co nejpodrobnější údaje o trojrozměrné struktuře těch makromolekul, které jsou hlavním cílem léku. V současné době existuje banka takových dat, včetně značného počtu enzymů a nukleových kyselin. K pokroku v tomto směru přispěla řada faktorů. Především byla zdokonalena rentgenová strukturní analýza a vyvinuta spektroskopie založená na nukleární magnetické rezonanci. Tato druhá metoda otevřela zásadně nové možnosti, protože umožnila stanovit trojrozměrnou strukturu látek v roztoku, tzn. v nekrystalickém stavu. Podstatným bodem byla skutečnost, že pomocí genového inženýrství bylo možné získat dostatečné množství substrátů pro podrobný chemický a fyzikálně chemický výzkum.
Pomocí dostupných dat o vlastnostech mnoha makromolekul je možné pomocí počítačů simulovat jejich strukturu. To dává jasnou představu o geometrii nejen celé molekuly, ale také jejích aktivních center interagujících s ligandy. Jsou zkoumány vlastnosti topografie povrchu substrátu, povaha jeho strukturních prvků a možné typy interatomárních interakcí s endogenními látkami nebo xenobiotiky. Na druhou stranu počítačové modelování molekul, použití grafických systémů a odpovídajících statistických metod umožňuje získat poměrně úplný obrázek o trojrozměrné struktuře farmakologických látek a rozložení jejich elektronických polí. Takové souhrnné informace o fyziologicky aktivních látkách a substrátu by měly usnadnit účinnou konstrukci potenciálních ligandů s vysokou komplementaritou a afinitou. Dosud se o takových příležitostech mohlo jen snít – nyní se stávají skutečností.
Počítačové modelování molekul je založeno na četných aproximacích a předpokladech. Předpokládá se tedy, že energie molekul je určena pouze souřadnicemi jejich atomů v prostoru. Ale ve skutečnosti molekuly nejsou nehybné a výpočty energie na počítači se provádějí na statických molekulách. V současné době se vyvíjejí metody molekulární dynamiky, které umožňují zohledňovat tepelný pohyb molekul, ale stále neexistují přístupy, které by spolehlivě zohledňovaly entropickou složku energie. Navíc v rozumném časovém rámci je možné spočítat životnost systému v řádu několika pikosekund.
Studium trojrozměrné struktury proteinů představuje velké potíže. Dosud neexistují žádné metody, které by dokázaly přesně předpovědět trojrozměrnou strukturu proteinu na základě jeho aminokyselinové sekvence. Ačkoli se používá metoda analogií, kdy se předpokládá, že identické aminokyselinové oblasti různých proteinů jsou složeny podobným způsobem. Experimentální získávání trojrozměrných snímků je zatíženo mnoha obtížemi: pro rentgenovou strukturní analýzu je nutná krystalizace proteinu (která je možná pouze u rozpustných proteinů) a možnosti nukleární magnetické rezonance jsou omezeny velikostí molekul proteinů. .
Role molekulárního modelování pro základní i aplikovaný výzkum v oblasti molekulární biologie a biochemie neustále roste. S tím souvisí zdokonalování matematického aparátu, růst produktivity výpočetní techniky a hromadění obrovského množství faktografického materiálu, který vyžaduje analýzu.
Simulace chemických reaktorů Slouží k predikci výsledků průběhu chemicko-technologických procesů za daných podmínek v zařízeních libovolné velikosti. Pokusy o provedení rozsáhlého přechodu z malého reaktoru na průmyslový reaktor s využitím fyzikálního modelování byly neúspěšné kvůli neslučitelnosti podmínek pro podobnost chemických a fyzikálních složek procesu (vliv fyzikálních faktorů na rychlost chemické přeměny v reaktorech různých velikostí se výrazně liší). Proto se pro přechod ve velkém měřítku používaly především empirické metody: procesy byly studovány v postupně rostoucích reaktorech (laboratorní, rozšířený, poloprovozní, poloprůmyslový reaktor, průmyslový reaktor).
Matematické modelování umožnilo zkoumat reaktor jako celek a provést přechod ve velkém měřítku. Proces v reaktoru sestává z velkého množství chemických a fyzikálních interakcí na různých strukturních úrovních - molekula, makrooblast, reaktorový prvek, reaktor. V souladu se strukturálními úrovněmi procesu je sestaven vícestupňový matematický model reaktoru. První úroveň (vlastní chemická transformace) odpovídá kinetickému modelu, jehož rovnice popisují závislost rychlosti reakce na koncentraci reaktantů, teplotě a tlaku v celém rozsahu jejich změn, pokrývající praktické podmínky procesu. Charakter dalších konstrukčních úrovní závisí na typu reaktoru. Například u reaktoru s pevným ložem katalyzátoru je druhou úrovní proces, který probíhá na jednom zrnu katalyzátoru, kdy je podstatný přenos látky a přenos tepla v porézním zrnu. Každá následující strukturní úroveň zahrnuje všechny předchozí jako součásti, například matematický popis procesu na jednom zrnu katalyzátoru zahrnuje jak přenosové rovnice, tak kinetické. Model třetí úrovně také obsahuje rovnice pro přenos hmoty, tepla a hybnosti v loži katalyzátoru atd. Modely reaktorů jiných typů (s fluidním ložem, sloupcového typu se zavěšeným katalyzátorem atd.) mají rovněž hierarchickou strukturu.
Pomocí matematického modelování se vyberou optimální podmínky pro proces, určí se potřebné množství katalyzátoru, velikost a tvar reaktoru, parametrická citlivost procesu na počáteční a okrajové podmínky, přechodové režimy a stabilita reaktoru. proces je také zkoumán. V řadě případů se nejprve provede teoretická optimalizace - určí se optimální podmínky, za kterých je výtěžek užitečného produktu největší, bez ohledu na to, zda je lze provést, a poté se ve druhé fázi navrhne technické řešení. zvolena tak, aby umožňovala nejlepší přístup k teoretickému optimálnímu režimu se zohledněním ekonomických a dalších ukazatelů. Pro realizaci nalezených režimů a normální provoz reaktoru je nutné zajistit rovnoměrné rozložení reakční směsi po průřezu reaktoru a úplnost promíchání proudů lišících se složením a teplotou. Tyto úlohy jsou řešeny fyzikálním (aerohydrodynamickým) modelováním zvoleného návrhu reaktoru.
Ke studiu různých procesů, ve kterých probíhají fázové a chemické přeměny, využívají metody termodynamického modelování.
Termodynamické modelování fázově-chemických přeměn je založeno na jedné straně na zákonech a metodách chemické termodynamiky, na druhé straně na matematickém aparátu pro řešení extrémních problémů. Plnohodnotná kombinace těchto dvou přístupů umožňuje implementovat výpočtovou metodu, která nemá zásadní omezení na charakter a komponentní charakter zkoumaných systémů.
Pro studium různých praktických a teoretických problémů spojených s fázovými a chemickými přeměnami je nutné hluboké a podrobné studium fyzikálně-chemické podstaty procesu, identifikace zákonitostí fázových a chemických přeměn probíhajících v tomto procesu, vlivu stavových parametrů (teplota, tlak, složení reakční směsi atd.).
Složitost většiny reálných fyzikálně-chemických procesů neumožňuje řešit popsané problémy výhradně experimentálně. Analýza možných přístupů ukazuje efektivitu přitahování moderních teorií a metod fyzikálně-chemického a matematického modelování a výpočtů pomocí termodynamických reprezentací. Pomocí těchto metod lze provést podrobné studium fázových a chemických přeměn.
Teoretické modelování
Role teoretického modelování ve vývoji chemické vědy je zvláště významná, protože svět atomů a molekul je skryt přímému pozorování výzkumníka. Poznání se proto provádí konstruováním modelů neviditelných objektů pomocí nepřímých dat.
Rýže. 2. Stavba a úprava modelů
Proces teoretického modelování, jak je uvedeno výše, se provádí ve fázích: sestavení modelu, studium modelu a extrapolace. V každé fázi lze identifikovat určité akce, které jsou nezbytné pro její realizaci. (Obrázek 2). Modely lze doplňovat, měnit a dokonce i nahrazovat novými modely. K takovým procesům dochází, když výzkumníci čelí novým faktům, které jsou v rozporu s vytvořeným modelem. Nový model je výsledkem přehodnocení rozporů mezi starým modelem a nově získanými daty.
Uvažujme o specifikách procesu poznávání v teoretickém modelování.
Ideální modelování je jednou z metod teoretického poznání. Základ teoretického modelování by tedy měly tvořit takové strukturální složky teoretických znalostí, jako je problém, hypotéza a teorie.
Po nashromáždění faktografického materiálu a jeho analýze je problém identifikován a formulován. Problém je forma teoretického poznání, jehož obsahem je něco, co člověk dosud nepoznalo, ale co je třeba poznat. Jinými slovy, je to znalost nevědomosti, otázka, která vyvstala v průběhu poznání a vyžaduje odpověď. Problémem není zamrzlá forma vědění, ale proces, který zahrnuje dva hlavní body (etapy pohybu vědění) – její formulaci a řešení. Správné odvození znalostí problému z předchozích faktů a zobecnění, schopnost správně položit problém je nezbytným předpokladem jeho úspěšného řešení. "Formulování problému je často zásadnější než jeho řešení, které může být pouze záležitostí matematického nebo experimentálního umění. Kladení nových otázek, rozvíjení nových možností, nahlížení na staré problémy z nového úhlu vyžaduje kreativní představivost a odráží skutečný úspěch ve vědě. "
V. Heisenberg poznamenal, že formulace a řešení vědeckých problémů vyžaduje: a) určitý systém pojmů, s jejichž pomocí bude badatel zaznamenávat určité jevy; b) systém metod zvolený s ohledem na cíle studia a povahu řešených problémů; c) spoléhání se na vědecké tradice, neboť podle Heisenberga „zásadní roli při výběru problému hraje tradice, průběh historického vývoje“, i když jistou důležitost mají samozřejmě zájmy a sklony samotného vědce.
Věda podle K. Poppera nezačíná pozorováním, ale problémy a její vývoj je přechodem od jednoho problému k druhému – od méně hlubokého k hlubšímu. Problémy podle něj vznikají buď jako důsledek rozporu v samostatné teorii, nebo když se střetnou dvě různé teorie, nebo v důsledku kolize teorie s pozorováním.
Vědecký problém je tedy vyjádřen v přítomnosti rozporuplné situace (působící ve formě opačných pozic), která vyžaduje odpovídající řešení. Rozhodující vliv na způsob kladení a řešení problému má za prvé povaha myšlení doby, ve které je problém formulován, a za druhé úroveň znalostí o těch objektech, které problém vyvstává. Každá historická epocha má své charakteristické podoby problémových situací.
K vyřešení identifikovaného problému vědec formuluje hypotézu. Hypotéza je forma teoretického poznání obsahující předpoklad formulovaný na základě řady faktů, jejichž skutečná hodnota je nejistá a je třeba ji prokázat. Hypotetické znalosti jsou pravděpodobné, nejsou spolehlivé a vyžadují ověření a zdůvodnění. V průběhu dokazování předložených hypotéz se některé z nich stávají pravdivou teorií, jiné jsou modifikovány, upřesňovány a konkretizovány, jiné jsou zahazovány a proměňují se v bludy, pokud test dává negativní výsledek. Formulace nové hypotézy se zpravidla opírá o výsledky testování staré, i když tyto výsledky byly negativní.
Takže například kvantová hypotéza předložená Planckem po testování se stala vědeckou teorií a hypotézy o existenci „kalorických“, „flogistonových“, „éterových“ atd., které nenašly potvrzení, byly vyvráceny. chyba. Fáze hypotézy prošel také D.I. Mendělejevův periodický zákon.
DI. Mendělejev věřil, že při organizaci cílevědomého, systematického studia jevů nic nemůže nahradit konstrukci hypotéz. "Oni," napsal velký ruský chemik, "jsou nezbytné pro vědu a zvláště pro její studium. Dávají harmonii a jednoduchost, které je obtížné dosáhnout bez jejich přijetí. Čas stát se pravdivým než žádný."
Hypotéza je podle Mendělejeva nezbytným prvkem přírodovědného poznání, který nutně zahrnuje: a) sběr, popis, systematizaci a studium faktů; b) vypracování hypotézy nebo předpokladů o kauzálním vztahu jevů; c) experimentální ověřování logických důsledků z hypotéz; d) převedení hypotéz na spolehlivé teorie nebo zahození dříve přijaté hypotézy a předložení nové. DI. Mendělejev jasně pochopil, že bez hypotézy nemůže existovat spolehlivá teorie: „Pozorování, zobrazování a popisování viditelného a podřízeného přímému pozorování – s pomocí smyslů můžeme během studia doufat, že se nejprve objeví hypotézy a teprve potom teorie toho, co je nyní nutné, dát do základu toho, co se studuje."
Hypotéza tedy může existovat pouze tehdy, pokud není v rozporu se spolehlivými fakty zkušenosti, jinak se stává pouhou fikcí. Ověřuje se (ověřuje) odpovídajícími experimentálními fakty (zejména experimentem), získávající charakter pravdy. Hypotéza je plodná, pokud může vést k novým poznatkům a novým metodám poznání, k vysvětlení široké škály jevů.
Hypotéza jako metoda rozvoje vědeckých a teoretických poznatků při její aplikaci prochází následujícími hlavními etapami.
1. Pokus o vysvětlení zkoumaného jevu na základě známých faktů a zákonitostí a teorií již existujících ve vědě. Pokud tento pokus selže, provede se další krok.
2. Předkládání dohadů, domněnek o příčinách a zákonitostech tohoto jevu, jeho vlastnostech, souvislostech a vztazích, o jeho vzniku a vývoji atd. V této fázi poznání je předkládaná pozice pravděpodobnou znalostí, která ještě nebyla logicky prokázána a není zkušenostmi natolik potvrzena, aby mohla být považována za spolehlivou. Nejčastěji je k vysvětlení stejného jevu předloženo několik předpokladů.
3. Vyhodnocení solidnosti, účinnosti předpokladů a výběr z jejich souboru nejpravděpodobnějších na základě výše uvedených podmínek platnosti hypotézy.
4. Nasazení předloženého předpokladu do uceleného systému poznání a deduktivní vyvození důsledků z něj s cílem jejich následného empirického ověření.
5. Experimentální, experimentální ověřování důsledků vyslovených z hypotézy. V důsledku tohoto testu hypotéza buď „přechází do hodnosti“ vědecké teorie, nebo je vyvrácena, „opouští vědeckou scénu“. Je však třeba mít na paměti, že empirické potvrzení důsledků hypotézy plně nezaručuje její pravdivost a vyvrácení jednoho z důsledků nesvědčí jednoznačně o její nepravdivosti jako celku. Tato situace je charakteristická zejména pro vědecké revoluce, kdy jsou zásadním způsobem narušeny základní pojmy a metody a objevují se zásadně nové myšlenky.
Rozhodujícím testem pravdivosti hypotézy je tedy nakonec praxe ve všech jejích podobách, ale určitou (pomocnou) roli při dokazování či vyvracení hypotetického poznání hraje i logické (teoretické) kritérium pravdivosti. Otestovaná a ověřená hypotéza se stává spolehlivou pravdou, stává se vědeckou teorií.
Různé strategie pro budování kinetických modelů komplexních reakcí
Chemická kinetika je věda o rychlostech chemických reakcí, o dynamickém chování reakčního systému na jeho cestě k chemické rovnováze. Tato oblast fyzikální chemie úzce souvisí se studiem mechanismů chemických reakcí, protože chemická kinetika je jednou z metod studia mechanismů a reakční mechanismus, jak se nyní ukázalo, je základem pro konstrukci adekvátní kinetický model.
Skončilo 20. století, století triumfálního rozvoje chemické kinetiky, zahrnující jak mikroúroveň elementárního aktu, tak makroúroveň vícestupňových procesů, vyznačujících se fenomenální složitostí mechanismů. Základy chemické kinetiky jako vědy položila počátkem století práce laureátů Nobelovy ceny J. Vant-Hoffa (1901), S. Arrhenia (1903), V. Ostwalda (1909) a také M. Bodensteina . Různé aspekty teorie elementárního aktu rozvinuli G. Eyring, M. Polyani, V. G. Levich a R. R. Dogonadze, laureáti Nobelovy ceny K. Fukui a R. Hoffman (1981), G. Taube (1983), R. Marcus ( 1992) a mnoho dalších badatelů. Teorie řetězových reakcí byla vytvořena pracemi M. Bodensteina, J. Christiansena a laureátů Nobelovy ceny N.N.Semenova a S.N.Hinshelwooda (1956), jejich studentů a následovníků.
Nobelovy ceny byly uděleny za metody a výsledky studia rychlých elementárních reakcí (M. Eigen, J. Porter, R. Norrish, 1967), jakož i za vývoj metod pro studium dynamiky elementárních aktů reakcí v plynné fázi ( D. Hershbach, J. Lee, J. Polani, 1986).
Vynikající výsledky byly získány v oblasti kinetiky homogenních a heterogenních katalytických reakcí. Zmiňujeme pouze teorii
kinetika heterogenních reakcí na nehomogenních površích (M.I. Temkin a S.Z. Roginsky), teorie kinetiky stacionárních reakcí Horiuti-Temkina, objev katalýzy paladiovými komplexy oxidačních přeměn olefinů (I.I. Moiseev, M.N. Vargaftik, Ya. Syrkin, Y.Smidt a další) a vytvoření teorie těchto procesů IIMoisejevem na základě podrobných kinetických studií (cena AP Karpinského, 1999).
Dvacáté století bylo korunováno pozoruhodným objevem nového oboru fyzikální chemie elementárního aktu, nazvaného „femtochemie“, a v roce 1999 udělením Nobelovy ceny za chemii americkému vědci, Egypťanovi A. Zewailovi „za studie přechodných stavů. femtosekundová metoda (10-15 sec.) laserová spektroskopie“. Byl dosažen limit měření rychlostí chemických reakcí. Bylo možné sledovat procesy probíhající při jedné vibraci atomů v chemické vazbě - 10 - 100 fs. Přechodový stav řady reakcí je zaznamenáván s rozlišením 0,1 Á podél reakční souřadnice s úplným spektrálním portrétem. Bylo dosaženo úrovně rozlišení sousedních energetických stavů ~ 10 –4 cm –1.
Všechny výsledky studia „nerovnovážné“ kinetiky chemických reakcí na mikroúrovni jsou nesmírně důležité pro doložení základních principů chemické kinetiky, ale zatím jsou málo použitelné pro řešení problémů na makroúrovni – zkoumání mechanismů komplexních reakcí v plynů, roztoků a na povrchu pevné látky za podmínek Maxwell-Boltzmannovy distribuce, tj. problémy "rovnovážné" kinetiky chemických reakcí. Pokud je prakticky vyřešen problém objasnění mechanismů a sestrojení kinetických modelů komplexních reakcí pro „rovnovážnou“ kinetiku reakcí radikálů a řetězců v plynné fázi (vzhledem k možnosti konstrukce maximálních mechanismů nebo reakčních sítí se známými rychlostními konstantami elementárních stupňů ), pak pro složité vícecestné procesy v řešeních a na povrchu řešení tento úkol teprve začíná. To je problém XXI. století.
Existují tři typy matematických modelů (matematický popis) složitých procesů. Stochastické modely využívají pravděpodobnostní reprezentace procesů ve výzkumném objektu. Funkce rozdělení pravděpodobnosti jsou vypočteny pro proměnné parametry modelu (koncentrace, teplota v případě chemických procesů). Tyto modely se v chemické kinetice stále používají zřídka, ale ukázaly se jako užitečné pro popis a modelování chování velkých systémů (chemické komplexy, chemické závody). Statistické modely se používají k popisu experimentu na pracovním výzkumném objektu. Je popsán vztah mezi hodnotami proměnných vstupujících do systému a proměnnými opouštějícími systém bez použití fyzikálně-chemických informací o procesech probíhajících v objektu (model černé skříňky). Matematickým popisem chování systému jsou obvykle rovnice ve formě polynomů. Pro zajištění statistické nezávislosti parametrů modelu se používá plánování experimentů (například ortogonální návrhy experimentů). Deterministické modely jsou založeny na zákonech fyzikálních a chemických procesů s určitou strukturou modelu. Teoreticky založené kinetické modely jsou právě takovými modely. Předmětem tohoto kurzu budou deterministické, strukturní, teoreticky podložené kinetické modely (KM) chemických procesů.
V matematickém modelování katalytického procesu existuje určitá hierarchie matematických modelů. Modely první úrovně jsou kinetické modely procesů na zrnu pevného katalyzátoru nebo v elementárním objemu kapalné fáze v homogenní reakci, nekomplikované procesy přenosu hmoty, tepla a hydrodynamických faktorů. Modely druhé úrovně v heterogenní katalýze uvažují procesy v loži katalyzátoru a modely třetí úrovně v homogenní a heterogenní katalýze jsou modely reaktoru jako celku, včetně všech přenosových procesů a struktury proudění. V tomto kurzu budou diskutovány modely první úrovně (CM). Takové modely jsou potřebné pro studium nových reakcí, pro optimalizaci katalytických procesů, pro výpočet průmyslových reaktorů (jako součásti matematického modelu reaktoru), pro vytvoření automatizovaných systémů řízení procesů.
O konceptu "reakčního mechanismu"
Konstrukce CM je tedy založena na mechanismu procesu, tzn. soubor elementárních stupňů vedoucích k přeměně výchozích činidel na konečné produkty reakcí a pro stejnou reakci (katalytickou či nekatalytickou) existuje konečný soubor mechanismů určovaných existujícím množstvím znalostí a paradigmat fungujících v chemie.
Například pro reakci nukleofilní substituce v aromatickém jádru ArX (nekatalytické, katalyzované kovovými komplexy nebo indukované přenosem elektronů z ArX na ArX) bylo stanoveno 8 mechanismů:



Bylo navrženo 13 jednocestných a 80 dvoucestných mechanismů jednoduché hydrogenační reakce ethylenu na kovových katalyzátorech. Jinými slovy, pro každý reakční systém (činidla, katalyzátor) existuje řada elementárních stupňů - reakční síť (maximální mechanismus), jejíž jednotlivé bloky jsou realizovány v závislosti na povaze katalyzátoru, podmínkách, substituentech v substrátu. a oxidační stav kovového katalyzátoru.
Na konci století byla nastíněna kombinace fyzikálně-chemických a formálně kinetických přístupů ke studiu mechanismů. Na mechanismus byl formulován pohled jako na jednotu dvou složek tohoto konceptu - topologické (strukturální) a chemické složky a jejich rovnost - nelze jednoznačně stanovit strukturu mechanismu (vztah elementárních stupňů) na základ pouze formálního kinetického popisu, tzv. "schéma mechanismu" a poté jej naplnit chemickým obsahem. V obecném případě je nemožné získat z kinetických experimentů informace potřebné pro správnou identifikaci schématu mechanismu bez specifikace tohoto mechanismu a bez stanovení příslušných úloh pro formální kinetickou metodu.
Všechny tyto okolnosti vyvolaly potřebu revidovat tradiční strategii budování CM.
Alternativní strategie budování CM
Tradiční postup pro konstrukci CM zahrnuje následující kroky:
Hlavní nevýhodou tohoto postupu (strategie) je absence algoritmů pro jednoznačné provádění všech fází. Protože experiment lze adekvátně popsat velkým množstvím matematických modelů (rovnic), musí mít výzkumník nějakou hypotézu o schématu mechanismu (o struktuře mechanismu) nebo o tvaru navržených rovnic. V tomto případě nastává výběr možných hypotéz (někdy intuitivní) až po provedení experimentu. Neexistuje žádný algoritmus pro přechod z matematického modelu na fyzikální model (zejména pro vícecestné reakce) (fáze (b)). Přechod na reakční mechanismus (stupeň (c)) je rovněž libovolný a není formalizován. Ve všech fázích této strategie existuje přirozená tendence získat alespoň jednu rovnici (a „diagram mechanismu“), která není v rozporu s experimentem, a velmi často zde není řeč o žádné diskriminaci sady hypotéz. . Naopak autoři takového schématu mechanismu začínají zakládat experimenty, aby dokázali mechanismus, který se autorovi zdá nejrozumnější. Přitom je již dávno stanoveno, že není možné prokázat žádnou hypotézu. Můžete přesvědčivě zahodit nepracovní hypotézy a prokázat souhlas s experimentem zbývajících hypotéz – souboru pracovních hypotéz. Vhodnost předložení souboru hypotéz a získání souboru pracovních hypotéz přesvědčivě doložil před více než 100 lety americký geolog T. Chamberlain.
Racionální strategie pro konstrukci CM je tedy jasným metodologicky podloženým hypoteticko-deduktivním logickým výzkumným schématem, podpořeným schopnostmi počítačů a efektivním softwarem. Podstata této strategie se odráží v posloupnosti jejích fází:

Diskriminace hypotéz může zahrnovat diskriminaci stádií, bloků stádií, jednotlivých mechanismů, spojení v reakcích s více drahami.
Typ CM (forma matematického popisu) závisí na vlastnostech mechanismu (lineární nebo nelineární), podmínkách procesu (stacionární, kvazistacionární, nestacionární), typu reaktoru (otevřený, uzavřený) a řadu přijatých předpokladů. Lineární mechanismus je mechanismus, jehož elementární stupně v dopředném a zpětném směru jsou lineární v meziproduktech - pouze jedna meziproduktová sloučenina je umístěna vlevo (nebo vpravo) od šipky v elementárním stupni. Pokud se jednoho meziproduktu účastní více než jeden meziprodukt (včetně 2 molekul jednoho meziproduktu), jsou fáze nelineární a mechanismus je nelineární.
Nejběžnějším typem CM jsou systémy diferenciálních rovnic, algebraicko-diferenciální nebo algebraické rovnice
![]() , (1)
, (1)
jehož pravá strana je vždy součinem matice stechiometrických koeficientů pro stupně mechanismu (transponované) sloupcovým vektorem rychlostí elementárních stupňů ( ![]() ). V případě lineárních mechanismů pro reakce ve stacionárních nebo kvazistacionárních podmínkách se pravá strana rovnice (1) transformuje na zlomkově-racionální rovnice pro rychlosti reaktantů (R i) nebo rychlosti pro cesty (R p) . V obecném případě se neřeší soustavy algebraických rovnic pro nelineární mechanismy a rovnice typu (1) nejsou redukovány na jednodušší zlomkový racionální tvar.
). V případě lineárních mechanismů pro reakce ve stacionárních nebo kvazistacionárních podmínkách se pravá strana rovnice (1) transformuje na zlomkově-racionální rovnice pro rychlosti reaktantů (R i) nebo rychlosti pro cesty (R p) . V obecném případě se neřeší soustavy algebraických rovnic pro nelineární mechanismy a rovnice typu (1) nejsou redukovány na jednodušší zlomkový racionální tvar.
V případě kinetiky na nehomogenních površích za stacionárních podmínek lze rychlost popsat i výkonovou rovnicí typu (2) (rovnice M.I. Temkina pro syntézu čpavku):
 (2)
(2)
Koeficient m = 0,5 v případě železného katalyzátoru, k + / k - = K je rovnovážná konstanta reakce
Vlastnosti kinetických modelů pro různé případy, metody pro odvození kinetických rovnic a metody pro konstrukci CM v rámci racionální strategie budou probrány v dalších částech kurzu. Zvládnutí tradiční strategie je předmětem domácího úkolu (kurzu).
Otázky pro sebeovládání
1) Uveďte nevýhody tradiční strategie.
2) Metodologické zdůvodnění racionální strategie.
3) Vyjmenujte hlavní fáze racionální strategie a vyjmenujte výhody této strategie ve všech fázích budování KM.
4) Vyjmenujte vlastnosti CM v případě lineárních a nelineárních mechanismů.
Literatura pro hlubší studium tématu
1. Schmid R., Sapunov VN, Neformální kinetika, M., Mir, 1985, 263 s. (Tradiční strategie).
2. Brook LG, Zeigarnik AV, Temkin ON, Valdes-Perez R., Metody hypotéz o mechanismech reakcí. Učebnice, Moskva: MITHT, 1999.
3. Temkin ON, Brook LG, Zeigarnik AV, Některé aspekty strategie pro studium mechanismů a konstrukci kinetických modelů komplexních reakcí, Kinetics and Catalysis, 1993, vol. 34, č. 3, str. 445–462.
4. Temkin ON, Problémy kinetiky komplexních reakcí, Ross. chemický časopis, 2000, v. 44, č. 4, s. 58–65.




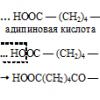
A katalýza katedry chemie a technologie základní organické syntézy MITHT pojmenovaná po M.V. Lomonosov. K vyslovení hypotéz o mechanismech syntézy kyseliny akrylové reakcí (6) v roztocích palladiových komplexů bylo použito 11 transformací: 1. 2. 3. 4. 5. 6. 7. 8. 9. 10. 11. programu ChemNet jsme získali reakci jedna...
Míra, synergický styl myšlení může být jakousi platformou pro otevřený tvůrčí dialog mezi vědci, mysliteli, umělci, kteří mají různé tvůrčí postoje a pohledy na svět. 2. Některé paradoxní důsledky synergetiky V synergetice vzniká mnoho nových paradoxních myšlenek, obrazů a reprezentací. Navíc z hlediska synergetiky může být ...





...; VCH3OH = 10 ml; 0:0:0 = 5:3:2. Důvody tohoto efektu budou zkoumány v průběhu dalšího výzkumu. 5. Patentová rešerše 5.1. Úvod Tato práce je věnována studiu podmínek pro vznik vibračního režimu při oxidativní karbonylaci alkynů v přítomnosti palladiových katalyzátorů. Tento proces je velmi zajímavý, protože dále umožní...





Aby podnik přežil v konkurenčním prostředí a úspěšně se rozvíjel na trhu, potřebuje jasně vypracovaný plán jak pro dlouhodobé, tak pro aktuální období. KAPITOLA 2. ANALÝZA POSTAVENÍ PODNIKU „AVTODOM-ATEX“ LLC NA TRHU SLUŽEB AUTOSERVISŮ 2.1 Stručná technická a ekonomická charakteristika podniku Podnik „Avtodom-Ateks“ byl založen na základě rozhodnutí účastníků z r. 23...
Ve fyzikální chemii se rychlost chemické reakce určuje podle rovnice:
kde dq- změna hmotnosti reaktantu, mol.
dt- přírůstek času, s.
PROTI Je mírou reakčního prostoru.
Rozlišujte mezi homogenními chemickými reakcemi, ve kterých jsou všechny zúčastněné látky ve stejné fázi (plyn nebo kapalina). Pro takové reakce je mírou reakčního prostoru objem a rozměrem rychlosti bude:.
Mezi látkami v různých fázích (plyn-pevná látka, plyn-kapalina, kapalina-kapalina, pevná látka-kapalina) probíhají heterogenní chemické reakce. V tomto případě je skutečná chemická reakce realizována na rozhraní, které je mírou reakčního prostoru.
U heterogenních reakcí je dimenze rychlosti různá:.
Změna hmotnosti reagujících látek má své vlastní znamení. U výchozích látek hmotnost v průběhu reakce klesá, změna hmotnosti má záporné znaménko a velikost rychlosti nabývá záporné hodnoty. U produktů chemické reakce se hmotnost zvětšuje, změna hmotnosti je kladná a kladné se předpokládá i znaménko rychlosti.
Zvažte jednoduchou chemickou reakci
Mezi jednoduché reakce patří ty, které se provádějí v jedné fázi a jdou do konce, tzn. jsou nevratné.
Pojďme určit rychlost takové chemické reakce. K tomu je nejprve nutné rozhodnout, pro kterou z látek bude rychlost reakce stanovena: vždyť A a B jsou výchozí látky a změna jejich hmotnosti je záporná a C je konečný produkt, a jeho hmotnost s časem roste. Kromě toho, ne všechny stechiometrické koeficienty v reakci jsou rovny jednotce, což znamená, že pokud je průtok A po určitou dobu roven 1 molu, průtok B za stejnou dobu bude 2 moly, a proto hodnoty rychlosti vypočítané ze změny hmotností A a B se budou lišit o polovinu.
Pro jednoduchou chemickou reakci lze navrhnout jedinou míru rychlosti, která se určí takto:
kde r i- rychlost i-tého účastníka reakce
S i Je stechiometrický koeficient i-tého účastníka reakce.
Stechiometrické koeficienty pro výchozí látky jsou považovány za kladné, pro reakční produkty jsou záporné.
Pokud reakce probíhají v izolované soustavě, která nevyměňuje hmotu s vnějším prostředím, pak pouze chemická reakce vede ke změně hmotností hmoty v soustavě a následně i jejich koncentrací. V takovém systému je jediným důvodem změny koncentrace S je chemická reakce. Pro tento konkrétní případ
Rychlost chemické reakce závisí na koncentracích obsažených látek a na teplotě.
kde k- rychlostní konstanta chemické reakce, C A, C B- koncentrace látek, n 1, n 2- objednávky příslušných látek. Tento výraz je ve fyzikální chemii známý jako zákon hromadného působení.
Čím vyšší jsou hodnoty koncentrace, tím vyšší je rychlost chemické reakce.
Objednat ( n) je stanoven experimentálně a je spojen s mechanismem chemické reakce. Pořadí může být celé číslo nebo zlomkové číslo, u některých látek existují i reakce nultého řádu. Pokud je objednávka i-tá látka je rovna nule, pak rychlost chemické reakce nezávisí na koncentraci této látky.
Rychlost chemické reakce závisí na teplotě. V souladu s Arrheniovým zákonem se rychlostní konstanta mění se změnou teploty:
kde A- preexponenciální faktor;
E- aktivační energie;
R- univerzální plynová konstanta, konstantní;
T- teplota.
Stejně jako velikost řádu reakce jsou pro konkrétní reakci experimentálně určeny hodnoty aktivační energie a preexponenciálního faktoru.
Pokud chemická reakce probíhá v heterogenním procesu, pak je její rychlost ovlivněna i procesem dodávání výchozích látek a odstraňování produktů ze zóny chemické reakce. Probíhá tedy složitý proces, ve kterém dochází k difúzním fázím (přísun, odběr) a kinetické fázi – vlastní chemické reakci. Rychlost celého procesu jako celku, pozorovaného v experimentu, je určena rychlostí nejpomalejšího stupně.
Ovlivněním rychlosti difúzního stupně procesu (míchání) tedy ovlivňujeme rychlost celého procesu jako celku. Tento vliv ovlivňuje hodnotu preexponenciálního faktoru A.
Většina chemických reakcí není jednoduchá (to znamená, že nejdou v jedné fázi a ne do konce) - složité chemické reakce:
a) AB - reverzibilní;
b) A -> B; В → С - po sobě jdoucí;
c) A -> B; A → C - paralelní.
Pro složitou chemickou reakci neexistuje jednotná míra rychlosti... Na rozdíl od jednoduchého zde můžeme mluvit o rychlosti tvorby a ničení každé chemikálie. Pokud tedy v systému probíhají chemické reakce a n látky pro každou z n látky mají svou vlastní hodnotu rychlosti.
Pro kteroukoli z látek je rychlost tvorby a zániku algebraickým součtem rychlostí všech stupňů za účasti této látky.